Fiscal 2000
(1) "Probing Molecular Symmetry in Liquids and Solutions by Polarization-resolved CARS (Coherent Anti-Stokes Raman Scattering) Spectroscopy"
Raman depolarization ratios carry useful information on molecular symmetry, which reflects sensitively the intermolecular interactions in liquids and solutions. We have developed a new non-linear Raman spectroscopic method, polarization-resolved CARS spectroscopy, which is capable of determining Raman depolarization ratios with unprecedented high accuracy of ±0.002, more than an order of magnitude better than the conventional spontaneous Raman method. In order to test the feasibility of the method, depolarization ratios were measured for two typical organic solvents, cyclohexane and 1,2-dichloroethane. For the two non-totally symmetric bands of liquid cyclohexane, r=0.749±0.002 and r=0.750±0.002 were obtained, in an excellent agreement with the theoretical value 0.75. This agreement indicates that the cyclohexane molecule retains D3d symmetry in neat liquid. The two totally symmetric bands of 1,2-dichloroethane, which were reported to have r=0.75 despite their symmetry, gave depolarization ratios of 0.746±0.003 and 0.742±0.003, values very little but significantly smaller than 0.75. The assignments of these bands to totally symmetric modes have thus been confirmed. The most recent experiments on aqueous MgSO4 solutions have suggested an ultrafast dynamic symmetry lowering of the sulfate ion from the tetrahedral symmetry, which is ascribed to the very short lifetime(∼ps) of the Mg2+-SO42- ion pair (Chem. Phys. Lett. 339, 351-356 (2001)).
(2) "A New Theoretical Approach to Solvent-induced Dynamic Polarization and Vibrational Dephasing of Molecules in Solution"
Elucidation of the solvent-solute interactions, especially the dynamic aspects of these interactions, is a key step for the microscopic understanding of chemical reactions in solution. We have previously shown that such solvent-solute interactions can be modeled as time-dependent solvent-induced dynamic polarization of the solute molecule(the dynamic polarization model). In order to substantiate this concept, we have developed a new theoretical approach in which a molecular dynamics (MD) simulation is used in conjunction with a molecular orbital (MO) theory to calculate the vibrational band shape of a dynamically polarizing solute molecule. The C=O stretch band of acetone in acetonitrile has been studied. By the MD simulation, we first obtained the instantaneous configurations of the solvent molecules. Then we calculated the instantaneous electric fields on acetone, which were combined with the vibratinal frequencies calculated by the finite-field MO method to obtain the Coulomb term of the instantaneous vibrational frequencies. The Lenard-Jones term was separately calculated classically. The instantaneous vibrational frequencies thus obtained were converted to the vibrational band shape using the Kubo theory. It has been found that this MD/MO approach reproduces the observed infrared C=O band shape very well without adjusting any MD parameters. We therefore believe that the obtained instantaneous MD configurations well represent the real solvent dynamics. The dynamic polarization behavior of acetone calculated from these MD configurations is shown in the Figure1. The present approach has opened way to study the solvent-solute interactions microscopically by analyzing the vibrational band shapes observed under various experimental conditions.
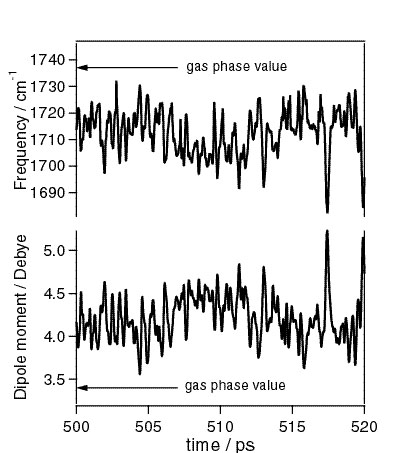
Fig. 1