Fiscal 1997
(1) "Picosecond energy dissipation of photoexcited molecules in solution : how a solute interacts with solvents"
Energy dissipation from excited states plays a crucial role in chemical reactions in solution. With the help of energy dissipation, the newly generated "hot" product molecule can release excess energy to reach the stable final state; otherwise it might go back to the energetically equivalent reaction complex or even to the reactant. We studied the process of excess-energy dissipation from photoexcited trans-stilbene in a number of different solvents by using picosecond time-resolved Raman spectroscopy. The 1570 cm-1 band of S1 trans-stilbene (C=C stretch) was found to be useful as an accurate temperature marker. The cooling-down kinetics in the time range of 0 to 70 ps was analyzed in terms of a diffusion equation of heat. The analysis led to a molecular model of the solute-solvent energy dissipation: the excess energy given to the solute by photoexcitation is shared with solvent molecules in the first solvation sphere in less than 3 ps. It is then dissipated to the bulk solvent in time-scales of 10-15 ps depending on the thermal diffusivity of the solvent (Fig. 1). The number of solvent molecules in the first solvation sphere is estimated to be five (V-V, V-R, V-T processes considered) to twelve (only V-R and V-T processes considered), from the temperature rise just after the photoexcitation.
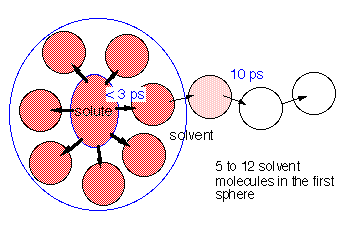
Fig. 1. Solvent-solute interaction and picosecond excess-energy dissipation.
(2) "New photoisomerization of diphenylacetylene as revealed by picosecond 2-D multiplex CARS spectroscopy"
Interaction among the closely-lying excited singlet states of diphenylacetylene has been a subject of considerable interest both experimentally and theoretically, as a model for strong electronic interactions. We used our picosecond 2-D CARS method to obtain structural information on these excited states. CARS spectra of both the S2 and S1 states were obtained with high S/N ratios. The central carbon-carbon stretch frequency of the S2 state was found to be 2009 cm-1. This frequency is indicative of a linear structure having a C≡C triple bond. No band was observed in the C≡C frequency region of the S1 CARS spectra. Isotopic substitution experiments and a two-mode normal coordinate analysis finally showed that the central carbon-carbon stretch frequency of the S1 state is ∼1570 cm-1. This value means that the central carbon-carbon bond of diphenylacetylene is a double bond (most probably in a bent form) rather than a triple bond. Diphenyl acetylene in the S1 state is not an acetylene. The S2 to S1 internal conversion of diphenylacetylene is known to have an activation energy of 1200 cm-1, which is unusual for a purely intramolecular relaxation process. It is most likely that the S2 to S1 internal conversion is a linear (S2) to bent (S1) isomerization that is accompanied by a large structural change (Fig. 2). This proposition is consistent with an empirical MO calculation and explains many curious photophysics of diphenyl acetylene reported previously.
Chem. Phys. Lett., 264, 551-555 (1997)
J. Phys. Chem. A 102, 2263-2269 (1998)
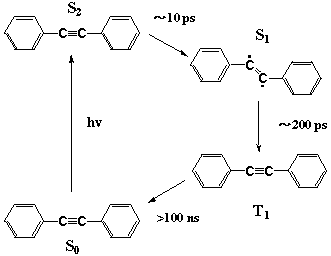
Fig. 2. Photoinduced dynamics of diphenylacetylene and new isomerization reaction