Fiscal 1999
(1) "Solvent-mediated Intermolecular Electron Transfer Reaction of Photoexcited Biphenyl in Alcohols"
The photoinduced electron transfer reaction plays crucial roles in many photophysical /photochemical/photobiological events of wide and general importance. We recently found and elucidated a new photoinduced electron transfer reaction in which the photoexcited biphenyl molecule undergoes intermolecular electron transfer to generate the cation and anion radicals. Using nanosecond time-resolved Raman spectroscopy, we have found that three transient species, the S1 state, the cation radical and the anion radical are formed after the photoexcitation of biphenyl in butanol. The S1 state is formed immediately after the photoexcitation and decays with a time constant of 9 ns. The signal of the cation radical reaches a maximum at 10 ns and decays slowly in the time range of a few hundred nanoseconds. The anion radical shows a delayed rise and its intensity reaches a maximum at 30 ns and decays faster than that of the cation radical. These temporal behaviors have been interpreted in terms of the solvent-mediated intermolecular electron transfer as shown in Scheme 1. The photogenerated S1 state spontaneously ejects an electron into the solvent to generate the cation radical and a solvated electron. The electron migrates in the solvent and encounters a ground-state biphenyl molecule to form the anion radical. The anion radical decays via the recombination with the cation radical and the cation radical decays via the recombination with electron/the cation radical. Thus, the cation radical lives longer than the anion. The kinetics of this electron transfer reaction depends markedly on the solvent properties; the spontaneous electron ejection from the S1 state is likely to be governed by the solvent polarity and its fluctuation, while the rate of electron migration may depend on the electron affinity and some particular motion of the solvent molecule. Elucidation of these points will one-step forward our understanding of the microscopic mechanism of chemical reaction in solution.
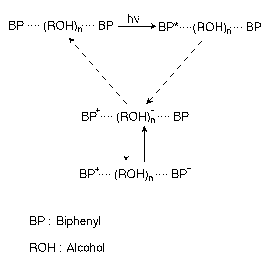
Scheme 1
(2) "The First Time-resolved Infrared Detection of A Carbene Intermediate in Solution at Ambient Temperature and Determination of the Singlet/triplet Energy Gap"
Nanosecond time-resolved infrared spectroscopy developed in our laboratory has been used to record the first time-resolved infrared spectrum of a carbene intermediate, 2-naphthylcarbomethoxycarbene in solution at ambient temperature. The work has been done in collaboration with Department of Chemistry, Johns Hopkins University , USA (Professor John Toscano). While studying the photochemistry of methyl 2-diazo-(2-naphthyl) acetateby time-resolved infrared spectroscopy, we have been able to observe two carbene species, the singlet and triplet 2-naphthylcarbomethoxycarbene. The two carbene intermediates decay with the same time constant indicating that they are in a thermal equilibrium. The decay rate agrees with the rise rate of the ketene product. This result means that the ketene is produced entirely from the carbene intermediate and not from the excited state of methyl 2-diazo-(2-naphthyl) acetate. We have also been able to determine the equilibrium constant and the free energy difference of the singlet and triplet carbenes in Freon-113 by referring to the intrinsic infrared intensity data provided by a low-temperature matrix study. A free energy difference of 0.2 + 0.1 kcal/mol has been obtained with the triplet carbene lower in energy. This is the first experimental determination of the singlet/triplet energy gap of a carbene intermediate in solution at ambient temperature. We have further confirmed the potential value of time-resolved infrared spectroscopy in structural and mechanistic studies of organic photochemistry in solution.