
時間分解赤外分光法とその生物物理化学的応用
濵口宏夫
1. はじめに
生体物質の構造変化のプロセスを、実時間かつin situで計測することができれば、生体機能を分子レベルで解明するうえで極めて有用な情報が得られる。生体物質の構造決定のための有力な手法として、X線回折法と核磁気共鳴(NMR)法がある。しかし、X線構造解析は結晶化が可能な試料にしか適用できない。また、NMR法は溶液中の分子構造について有用な情報を与えるものの、高速構造変化を追跡することができない。そこで最後の切り札として登場するのが、本稿の主題である時間分解赤外分光法である(と筆者は信じている)。周知のように、赤外線吸収やラマン散乱によって観測される分子の振動スペクトルは、「分子の指紋」と呼ばれ、分子構造の変化を鋭敏に反映する。振動スペクトルから得られる構造情報は、X線回折やNMRで得られるものほど精密ではないが、生体機能の機構を解明しようという目的においては十分本質的であり得る。
時間分解赤外分光の歴史は1940年代に遡るが1)、実用的な分光手法として脚光を浴びるようになったのは、1990年以降である。現在の発展の方向にはパルスレーザーによるフェムト/ピコ秒領域での赤外レーザー分光と赤外白色光源を用いたナノ/マイクロ/ミリ秒の領域の時間分解赤外分光の2つの流れがある。フェムト/ピコ秒領域では、クロモフォアの光物理化学的性質が主たる研究対象となるのに対し、ナノ/マイクロ/ミリ秒の領域では、蛋白質の高次構造変化や物質輸送など、生体機能の発現の現場での構造変化が捉えられる可能性がある。現在盛んに研究が行われているCOヘモグロビンの光解離や、バクテリオロドプシン光化学反応サイクルなどの系でも、興味の中心がフェムト/ピコ秒の初期過程から、ナノ/マイクロ/ミリ秒の遅い過程に移って来ている。生体機能の解明にとってに本質的で新しい情報は、ナノ秒より遅い時間領域の研究から得られる可能性が高いと思われる。本稿では、このような観点から、ナノ/マイクロ/ミリ秒領域での時間分解赤外分光の現状について、筆者らが開発した測定システムを例にして解説する。フェムト/ピコ秒の領域の研究については、最近の総説をあげるにとどめる2)。
2. ナノ/マイクロ秒時間分解赤外分光
この時間領域での時間分解赤外分光の技術は、すでにかなり成熟しており、過去1~2年の間に市販品も登場した。現在2つの異なる方式が併存している。第1は筆者らが開発してきた分散型の分光器を用いる方式であり3)、第2はドイツのSiebertらが中心になって開発してきたステップ-スキャンFT-IR分光計を用いる方式である4)。いずれの方式でも、赤外白色光源を用い、検出器にAC結合した広帯域増幅器により、光、電場などの外部摂動により誘起された透過赤外光強度の変化分のみを検出する。これら2つの方式には一長一短がある。広い波数範囲で時間分解赤外スペクトルを観測する必要があるときにはFT-IR法が、狭い波数範囲のキーバンドに注目してそのダイナミクスを詳しく調べたいときには分散法が有利である。また、FT-IR法の本質的な弱点である試料の経時変化に対する過敏性にも留意しておく必要がある。例えば、スペクトルの掃引中に何らかの理由で試料の交換が必要となった場合、分散方式ではその時点までに観測したデータはそのまま有効で、残りの部分のみを続けて測定すれば良いが、FT方式では最初からやり直さなければならない。実際の応用に際しては、これらの長所、短所を勘案して、どちらの方式を採用するかを決める必要がある。
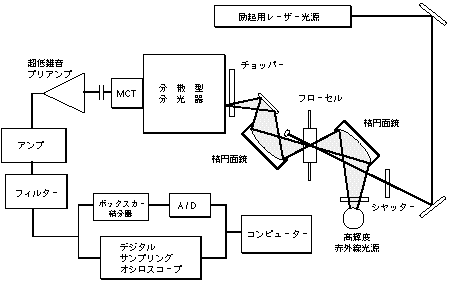
図1: 分散型分光器を用いた時間分解赤外分光装置
図1は筆者らが開発した分散型時間分解赤外分光計のブロック図である3)。市販の分散型赤外分光計の光源部、試料部、検出部を以下のように改造している。光源部には、通常のグローバー光源に代えてMoSi2高輝度赤外光源を使用している。試料部には一対の楕円面鏡からなるビームコンデンサーを置き、赤外光のビームサイズを直径数mmにしぼりこむ。これにより、試料の光励起に十分必要な励起レーザーの光子密度を確保している。検出部は、MCT(水銀-カドミウム-テルル)半導体赤外検出器とそれにAC結合した前置増幅器、フィルター、デジタルオシロスコープからなり、MCT検出器のAC出力をデジタルオシロスコープで時間分解計測する。MCT検出器は、測定対象に応じて光起電力型と光導電型を使い分けている。信号の処理、分光器の掃引、シャッターの開閉などをパーソナルコンピューターで制御している。各部分の詳細(実際に用いた機種等)については文献3を参照して頂くことにして、この装置の大まかな性能を以下に列挙しておく。測定波数範: 4000-700cm-1. 時間分解能: 50ナノ秒(光起電力型MCT). 検出可能最小吸光度変化: 3×10-7(光導電型MCT), 1×10-5(光起電力型MCT).
つぎに、全-トランスレチナールの光異性化反応を時間分解赤外分光で追跡した実験5)について述べる。
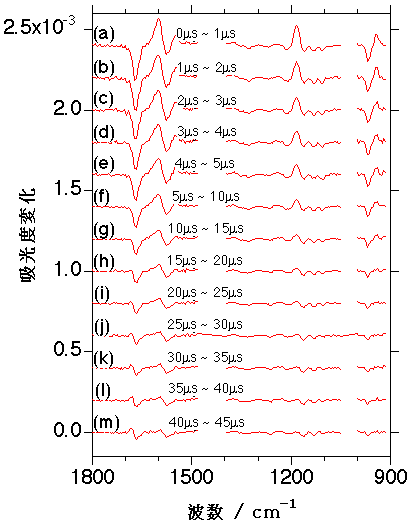
図2: シクロヘキサン中の全-トランスレチナールを光励起したときの時間分解赤外吸収スペクトル。上向きのバンドは光励起によって生成した励起3重項状態の全-トランスレチナールおよび光異性化反応によって生成した13-シスレチナールと9-シスレチナールに、下向きのバンドは光励起によって減少した基底状態の全-トランスレチナールに帰属される。
図2は、幅約7ns、波長349nmの紫外レーザーパルスで光励起したときの、全-トランスレチナールのシクロヘキサン溶液の時間分解赤外差スペクトルである。AC結合方式を用いているために、得られるスペクトルは差スペクトルの形になっている。下向きのピークは光励起によって分子数が減少した基底電子状態の全-トランスレチナールの赤外吸収バンドに、上向きのピークは光励起によって生成した励起電子状態の全-トランスレチナールや異性化反応生成物の赤外吸収バンドに対応している。図2のスペクトルから光励起された全-トランスレチナールが励起1重項状態を経由して、13-シスレチナールと9-シスレチナールに約3:1の比で異性化することがわかった。最低励起3重項状態を経由したモノシスレチナールから全-トランスレチナールへの光異性化過程はすでに良く知られていたが、励起1重項状態を経由した全-トランスレチナールから13-シスレチナールと9-シスレチナールへの異性化過程の存在は、時間分解赤外分光の研究で初めて明らかとなった。
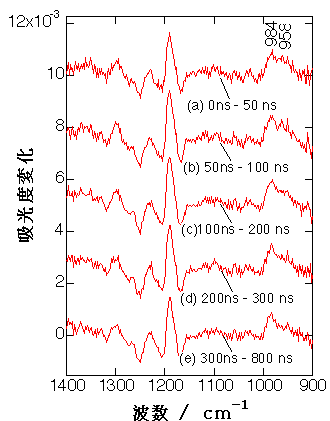
図3: バクテリオロドプシンの光化学反応サイクルのナノ秒時間分解赤外光。950~1000cm-1領域にK中間体からKL中間体への変換が観測されている。
図3は、バクテリオロドプシンの光化学反応サイクルを、分散型時間分解赤外分光法によって追跡したもので、京都大学の前田章夫教授らのグループとの共同研究によるものである6)。バクテリオロドプシンのクロモフォアはシッフ塩基結合した全-トランスレチナールで、光の吸収によりそれが13-シスレチナールに異性化し、プロトンポンプの光化学反応サイクルがスタートする。このスペクトルの解析から、光化学反応サイクル中に従来その存在が疑問視されていたKL中間体が確かに存在すること、またKL中間体とL中間体が平衡にあるなど新しい知見が得られた。現在、マイクロ秒からミリ秒の時間領域のスペクトル変化の解析が進行中である。
3. ミリ秒マルチチャンネルFT-IR分光法
前項で述べた分散型時間分解赤外分光法は、繰り返し(>100Hz)可能な過渡現象に関しては極めて有効である。しかし、繰り返しが不可能であるか、繰り返しが可能としても速度を低くおさえる必要がある、興味ある過渡現象も少なからず存在する。このような過渡現象を追跡するためには、また別の工夫が必要である。 筆者らは、単発の過渡現象にも適用可能な時間分解赤外分光装置として、マルチチャンネルFT-IR分光計を試作した(図4)7)。マルチチャンネルFT-IR分光計では通常のFT-IR分光計で用いられるマイケルソン型の干渉計とは異なる原理の干渉計が用いられる。マイケルソン干渉計では、ビームスプリッターにより2分された光束が、可動鏡の位置によって決まる位相差を持って検出器上で重ね合わされる。その結果、可動鏡の位置の関数としてインターフェログラムが得られる。マルチチャンネルFT-IR分光計では、特殊な干渉光学系を用いて、インターフェログラムを空間上に形成し、それをマルチチャンネル赤外検出器によって同時検出する。マルチチャンネルFT方式は、可動鏡を掃引する必要がないために、時間領域での測定における帯域の制約がなく、時間分解測定に適している。もし超高速の赤外白色パルスをこのマルチチャンネルFT-IR分光計と組み合わせることができれば、単一過渡現象の超高速時間分解赤外分光も原理的には可能である。通常の赤外白色光源と組み合わせた時の時間分解能は、マルチチャンネル赤外検出器の読みだし時間で決まる。
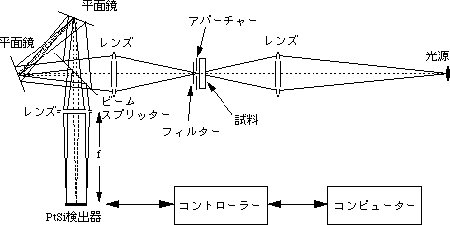
図4: マルチチャンネルフーリエ変換赤外分光装置
図4のシステムでは、三角光路コモンパスと呼ばれる干渉光学系を採用し、検出器として1024チャンネルのtSiアレイ検出器を用いている。データの読み出し時間は最速5ミリ秒で、単発過渡現象にともなう赤外吸収スペクト変化を、5ミリ秒毎に連続して観測することができる。PtSiアレイ検出器の感度特性のために、測定可能な波数範囲が500-2500cm-1に限られている。そのため、構造情報に富む指紋領域の測定は現在のところ不可能である。
この装置をレーザー誘起温度ジャンプ法と組み合わせ、脂肪酸結晶の融解と再結晶化の過程を追跡する実験を行った。図5にその結果を示す。
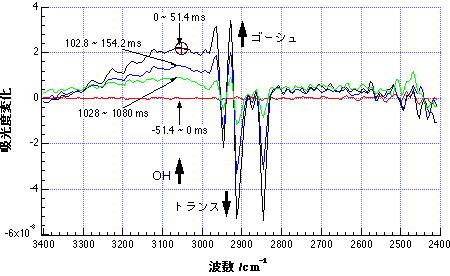
図5: 温度ジャンプ法と時間分解赤外分光の組み合わせによるデカン酸配向結晶の融解および再結晶化過程の追跡。上向きの鋭いバンドは、融解によって生成したゴーシュ構造に、上向きの幅広いバンドは、融解による配向の乱れによって観測されたOH伸縮バンドに、下向きのバンドは融解によって減少したトランス構造にそれぞれ帰属される。
試料は、KRS-5とCaF2の窓板の間に生成したデカン酸の配向結晶である。この試料に、Nd:YAGレーザーの第2高調波(532nm)のナノ秒パルスをCaF2側から照射すると、レーザー光の吸収によりKRS-5板が瞬間的に熱せられ、続いて熱伝導によりデカン酸の結晶の温度が上昇する(温度ジャンプ)。試料の温度を融点のわずかに下に調節しておくと、レーザー照射によって瞬時(ミリ秒に比較して極めて短い時間内)にデカン酸の結晶を融解させることができる。レーザーのパワーを調節して、試料に注入される熱エネルギーを適切に選ぶと、配向結晶の表層100分子層程度のみを融解させることができる。融解した表層の分子は、時間が経過し温度が下がるにしたがって元の配向を回復して再結晶化する。結晶の配向性を利用し、デカン酸のOH伸縮振動のバンドの遷移双極子モーメントと垂直方向に偏光した赤外光を用いると、配向が乱れた液体状態のデカン酸分子のOH伸縮振動バンドのみを選択的に検出することができる。
図5の時間分解赤外スペクトルから、融解後200ミリ秒でデカン酸の液体/結晶系は平衡状態に到達し、ゴーシュ構造がトランス構造に戻る速度と、液体中の分子が配向を回復して結晶化する速度とが等しくなることがわかった。このことから、結晶化は、液体中のトランス構造を経由してのみ進行することが示唆された。
マルチチャンネルFT-IR分光計による単発過渡現象の時間分解測定は、今ようやく端緒が開かれたばかりで、ここにあげたデカン酸結晶の融解と再結晶化の過程を追跡した実験が最初かつ唯一の例である。筆者は今後、この手法を様々な系へ応用したいと考えている。例えば蛋白質の熱変性過程の研究などは、図5の実験とそっくり同じ道具立てで可能であるし、光励起pHジャンプやイオン濃度ジャンプと組み合わせれば、生体機能と直接に関連した興味ある実験がいろいろと構築できそうである。
4. おわりに
筆者は生物に関して全くの素人であるが、いくつかの共同研究を通じて、物理化学系としての生体の構造と機能の面白さを学んで来たつもりである。今後も時間分解赤外分光法をはじめ、自らが開発した新しい分光手法を、興味ある生体系に応用する共同研究を積極的に推進したいと思っている。本稿がそのような共同研究のきっかけとなれば幸いである。
最後に、本稿で紹介した研究に寄与された多くの共同研究者諸氏に感謝する。
文献
- Baker, E. B., Robb, C. C. (1943) Rev. Sci. Instru. 14, 362.
- Locke, B., Diller, R., Hochstrasser, R. M. (1993) Advances in Spectroscopy (Clark, R.J.H., Hester, R.E., eds.) 21, 1-47, Wiley, Chichester, U.K.
- Yuzawa, T., kato, C., George, M. W., Hamaguchi, H. (1994) Appl. Spectrosc. 48, 684-690.
- Weidlich, O., Siebert, F. (1993) Appl. Spectrosc. 47, 1394.
- Yuzawa, T., Hamaguchi, H. (1995) J. Mol. Struct., 352/353, 489-495.
- Sasaki, J., Yuzawa, T., Kandori, H., Maeda, A., Hamaguchi, H. (1995) Biophys. J., 68, 2073-2080.
- Hashimoto, M., Hamaguchi, H. (1996) Appl. Specrosc. 50, 1030-1033.