
新しい振動分光学的手法の開発と応用
はじめに
筆者は過去35年にわたって、ラマン散乱や赤外線吸収を基礎とした新しい振動分光学的手法の開発と応用の研究に携わって来た。そして今、振動分光学の奥行きの深さを改めて実感するとともに、そのさらなる発展を強く予感している。
表1に示すように、これまでに筆者が開発、製作に関与した振動分光装置は20を越える。内訳はラマン分光が最も多く10種、非線形ラマン分光が9種、赤外分光が5種である。これらの装置のうち、筆者が実際に製作に携わったのはラマン分光の最初6種のみであり、その他の装置は様々な立場の共同研究者の手によるものである。この場を借りてこれら共同研究者の方々に厚くお礼を申し上げたい。
新しい手法を開発しようとするとき、新しい高価な道具を自在に使えるということは、極めて有利な条件である。その意味で筆者は大変恵まれた環境に置かれてきた。1981年から5年間の特別推進研究(代表者田隅三生教授)、1990年から5年間の神奈川科学技術アカデミー「極限分子計測」プロジェクト、そして2003年から5年間の学術創成研究と3つの大型研究費により、最新の道具を用いて装置製作に挑戦する機会を与えられたからである。これらのプロジェクトが、それぞれナノ秒、ピコ秒、フェムト秒レーザーの技術革新とうまく位相が合ったことも幸運であった。これらの研究費に基づく成果はしたがって、「単にレーザー技術開発の波に乗り、高価なレーザー装置を振動分光に導入した結果に過ぎない」のではないかと常に自戒の種としている。
| Raman Spectroscopy | High-resolution Raman
|
|---|---|
UV-VIS resonance Raman
| |
Compact gas Raman
| |
Matrix isolated Raman
| |
Nanosecond time-resolved Raman
| |
Multichannel cw Raman
| |
Picosecond time-resolved Raman
| |
NIR-excited Raman
| |
Low-frequency Raman
| |
Direct Raman imaging
| |
| Non-linear Raman Spectroscopy | Inverse Raman
|
Partially CARS
| |
Picosecond time-frequency 2-D CARS
| |
Polarization sensitive CARS
| |
Picosecond OKE gated CARS
| |
Femtosecond OKE
| |
Broadband multiplex CARS microspectroscopy
| |
CARS spatial distribution
| |
Hyper-Raman microspectroscopy
| |
| Infrared (IR) Spectroscopy | Microsecond Time-resolved IR
|
Nanosecond Time-resolved IR
| |
Millisecond multiplex FT-IR
| |
Infrared electroabsorption
| |
Sub-picosecond time-resolved IR
|
上記3つの大型プロジェクトでは、振動分光に時間分解能と空間分解能を付与し、その適用範囲を拡大することを目指した。短寿命反応中間体から生きた単一細胞まで、物質の階層構造を縦貫して広く応用されるに至った現代の振動分光では、その目標は相当程度達成されたと考えられる。これらのプロジェクトの看板である「時空間分解振動分光」については、これまでも機会があるごとに紹介してきた。そこで本講演では、高度に専門的であり(したがって分光学者以外には難しく)紹介する機会が少ない、しかし筆者が強い思い入れを持つ、振動分光3種、偏光分解CARS分光、電場変調赤外分光、CARS信号空間分布測定、を紹介することにしたい。これらの研究が単なるレーザーの高性能化の産物ではなく、将来の振動分光のブレークスルーにつながる可能性が高い「深さ」を持っていると考えるからである。
偏光分解CARS分光
CARS(Coherent Anti-Stokes Raman Scattering)には、2つの入射光(角振動数&omega1およびω2、波数ベクトルk1およびk2)と信号光(ωCARS、kCARS)が関与する(図1)。角振動数差ω1-ω2が試料のラマン活性モードの角振動数Ωと一致すると、ラマン共鳴によって3次の非線形感受率が増大し、角振動数2ω1-ω2、波数ベクトルk=2k1-k2を持つCARS光がビーム状に発生する(後出図8も参照)。
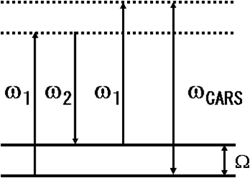
図1.CARS過程のエネルギーダイアグラム
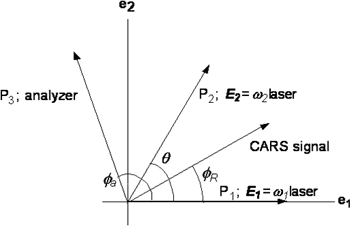
図2.偏光分解CARSにおける偏光設定
偏光分解CARS分光では、ω1光とω2光の電場E1とE2の偏光面が角度θをなすように固定し、検光子通してCARS信号光を観測する(図2)。このとき信号光とE1の偏光面のなす角度φRはtanφR=ρtanθで与えられる。ここでρはラマン散乱の偏光解消度である。検光子を回しながらCARS信号光を観測すると、ある特定の角度で特定の偏光解消度を持つバンドが消失する。θ=60°、ρ=0.75のとき、この角度は142°である。図3は、1,2-ジクロロエタンのCH2はさみ振動のバンドの偏光分解CARSスペクトルである。1443cm-1はトランス体、1429cm-1はゴーシュ体のバンドである。142°付近で信号強度が小さくなることがわかる。バンド形のフィッティング解析からCARS強度がゼロになる角度は1443cm-1で141.5°、1429cm-1で141.6°であることがわり、偏光解消度がそれぞれ0.742±0.003、0.746±0.003と決定された。これらのバンドはいずれも全対称振動に帰属されていたが、偏光解消度が0.75に近いことから非全対称振動の可能性も指摘されていた。偏光分解CARSの結果により、ρ<0.75が確定し永年にわたる問題が解決された。
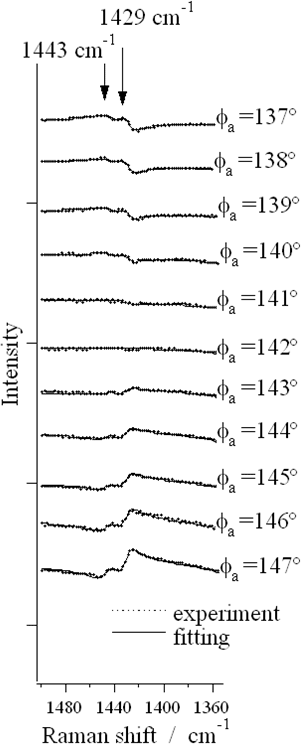
図3.1,2-ジクロロエタンの偏光分解CARSスペクトル
電場変調赤外分光
電場変調赤外分光は、外部から印加した電場によって誘起される赤外吸収の変化から、通常の赤外分光では得られない高次の分子情報を得ようとする手法である(図4)。その重要性にも拘らず、技術上の困難があり、溶液・液体試料の測定は3000cm-1の高波数領域に限られていた。筆者等は、分散型分光器をAC結合検出方式と組み合わせた方式により、10-8の微小な吸光度変化を検出することができる装置を開発した[2]。その結果、指紋領域での溶液・液体試料の電場変調赤外分光が可能となった。
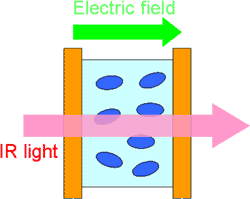
図4.電場変調赤外分光の概念図
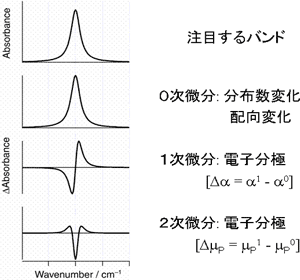
図5.電場変調赤外スペクトルの波形
電場変調赤外吸収スペクトルには、赤外吸収バンドの0次微分、1次微分、2次微分の波形が現われる(図5)。実際の電場変調赤外吸収スペクトルはこれらの重ね合わせの複雑な波形を示すが、入射赤外光と印加電場のなす角χへの依存性を調べることによって分離することができる。図6は、CCl4/CH3CN混合溶媒中のパラ-ニトロアニリンの電場変調赤外スペクトルのχ依存性を調べた結果である[3]。この解析から溶液中でパラ-ニトロアニリンPNAはCH3CNと2種類の溶媒和構造、1:1構造(図7-a)および1:2構造(図7-b)をつくることがわかった。
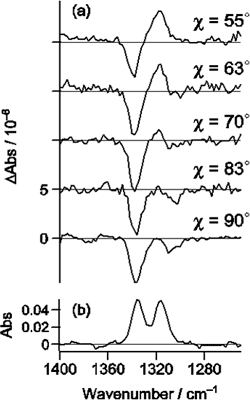
図6.pNAの電場変調スペクトルのχ依存性
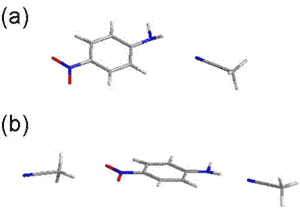
図7.pNA/CH3CNの2種の溶媒和構造
CARS信号の空間分布測定
CARSの位相整合条件は、試料媒質が光学的に均一で、&omega1、ω2、およびωCARS光の相互作用長Lが無限に長いとき厳密に成立し、その場合、信号光は特定の方向にのみビーム状に放出される。液体や溶液など試料媒質が完全には均一でなく、相互作用長Lに制限があると、位相整合条件が緩和され、CARS信号が空間分布を持つようになる(図8)。このCARS信号の空間分布を観測、解析することにより、液体や溶液中の局所構造についての定量的な情報が得られる可能性がある[4]。
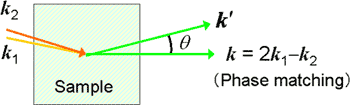
図8. CARSの位相整合と信号の空間分布
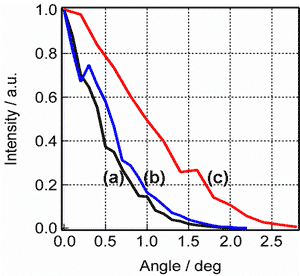
図9. エタノール40%水溶液のCARS信号空間分布の時間依存性
図9はエタノールの40%水溶液からのCARS信号の空間分布を測定した結果である。混合直後の空間分布(b)は、純エタノール(a)とほぼ同一であった。しかし、それを2週間放置すると、空間分布が有意に広がり、(c)のようになった。混合直後と2週間後では、アルコールと水の微視的混合状態が変化し、結果としてCARSの位相整合条件がより緩和されたものと考えられる。ポリスチレンビーズのモデル系での実験との対比から、この経時変化は、混合直後に存在するエタノールクラスターが、溶媒和により徐々に小さくなって行くことに対応しているものと考えられる。一方、混合直後と2週間後の通常のラマンスペクトルは寸分違わず一致しており、この変化は検出されなかった。ウイスキーが年を経るとともにまろやかな味に熟成されて行くプロセスが、CARS信号空間分布測定により、分光学的に解明されることになるかも知れない。
終わりに
振動分光学の間口は広がり、底知れず深い奥行きも垣間見えてきた。基礎、応用を問わずブレークスルーの可能性がかつてなく高まっているように筆者には感じられる。
文献
- Y. Saito, T. Ishibashi and H. Hamaguchi, J. Raman Spectrosc. 31, 725 (2000).
- H. Hiramatsu and H. Hamaguchi, Appl. Spectrosc. 58, 355 (2004).
- S. Shigeto, H. Hiramatsu and H. Hamaguchi, J. Phys. Chem., A 110, 3738 (2006).
- S. Shigeto and H. Hamaguchi, Chem. Phys. Lett. 417, 149 (2006).