
時間分解振動分光法
濵口宏夫
1 はじめに
赤外線吸収スペクトルやラマン散乱スペクトルに基づく振動分光法が、分子構造解析の手段として重要な役割を果たしていることは言うまでもない。近年のレーザー技術、光エレクトロニクス技術の進歩により、主として光によって生成する短寿命過渡分子種(ピコ秒からマイクロ秒の時間しか存在しない電子・振動励起分子、準安定中間体分子、ラジカルなど)の振動スペクトルを、時々刻々、時間の関数として測定することが可能となった。時間分解振動分光法の誕生である。高速時間分解能の獲得により、振動分光法の重要性はより一層高まった。他の有力な分子構造解析手法である磁気共鳴法や回折法では、このような高速の時間分解能が得られないからである。本稿では、分析化学への応用を念頭に置きながら、時間分解振動分光法の原理、装置、そして応用について解説する。 この手法が「進歩総説」でとりあげられるのは今回が最初であるので、これまでの発展経過のあらましを述べるとともに、解説の対象とする期間を過去約5年(1993年から1998年)と長めに設定した。また紙面の制約から、文献の引用は代表的と思われるもののみに限定したので、あらかじめお断りしておく。
2 時間分解振動分光法の発展経過のあらまし
時間分解振動分光法の発展の歴史は、1940年代のいわゆる「ラピッドスキャン」赤外分光の試み1)にまで遡ることができる。赤外分光器の鏡を高速で回転し、スペクトルをオシログラムとして記録しようという試みであったが、実用には至らなかった。本格的な応用という意味では、1970年代後半のマイクロ秒時間分解ラマン分光の実験の成功2)をその端緒とするのが妥当であろう。1980年代に入り、安定なナノ秒Q-スイッチNd:YAGレーザーが市販されるようになると、ナノ秒領域の時間分解ラマン分光が急速に発展した。数多くの短寿命過渡種のラマンスペクトルが測定され、それまで得ることのできなかった貴重な構造情報がもたらされた。1980年代の後半までには、ナノ秒時間分解ラマン分光の測定手法が確立された(総説3およびその引用文献参照)。1990年代に入ると、モード同期ピコ秒レーザの増幅技術が進歩し、ピコ秒領域での時間分解ラマン分光の試みが本格的化した。1993年にはNd:YAG再生増幅器を励起光源とする色素増幅器を用いた、フーリエ変換限界のピコ秒時間分解ラマン分光装置の製作が報告された。その後、多くのグループが再生増幅器を用いた方式を踏襲し現在に至っている。第3章でピコ秒時間分解ラマン分光とその応用について述べる。
一方、時間分解赤外分光の発展は、ラマン分光に対してやや遅れをとった。1980年代にいくつかのグループにより先駆的研究が行われていたが(総説4およびその引用文献参照)、論文数がラマン分光と肩を並べるようになったのは、1990年代に入ってからのことである。時間分解赤外分光の研究には、おおまかにいって2つの流れがある。第一の流れは、グローバーなどの通常光源を用い、電気的なゲート検出によりナノ秒からマイクロ秒の時間分解能を得るものである。現在では、分散型の分光器を用いる方式5)と、ステップ掃引FT方式6)の装置がすでに市販されている。第4章でナノ・マイクロ秒時間分解赤外分光とその応用について述べる。第二の流れは、超高速パルスレーザーを用いてピコ秒の時間分解能を達成するものである。ピコ秒の赤外パルスを検索光として用いる方式と、ピコ秒の紫外・可視パルスにより連続発振の赤外検索光にゲートをかける方式が行われている。第5章で、ピコ秒時間分解赤外分光の現状に触れる。
3 ピコ秒時間分解ラマン分光とその応用
3.1 フーリエ変換限界
レーザーパルスのパルス(時間)幅とスペクトル幅を同時に無限に狭くすることはできない。図1に、モード同期色素レーザーから最も普通に得られるsech2型の波形を持つレーザーパルスについて、同時に実現できるパルス幅Dtとスペクトル幅Dnの限界(フーリエ変換限界)を表わす曲線を示す。
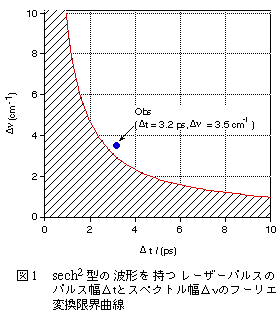
図1
この曲線からわかるように、1psのパルス幅を持つレーザーパルスは、10cm-1以上のスペクトル幅を持つ。したがって、このパルスを用いて時間分解ラマン測定を行うと、1ps程度の時間分解能を実現することが可能であるが、10cm-1より高いスペクトル分解能を得ることはできない。逆に1cm-1程度のスペクトル分解能を要求すると、時間分解能は10psまで低下することになる。図中に青い円●で示した点は、Iwataらにより用いられたレーザーシステム7)での実測値であり、パルス幅3.2ps、スペクトル幅3.5cm-1に対応する。これらの値はフーリエ変換限界内で、パルス幅とスペクトル幅を時間分解ラマン分光の目的に最適化したものである。
3.2 装置および実験法
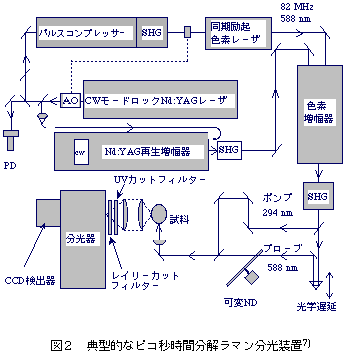
図2
典型的なピコ秒時間分解ラマン分光装置7)のブロック図を図2に示す。モード同期Nd:YAGレーザーからのパルス幅65psの出力をパルス圧縮器により幅5psに圧縮し、その倍波により同期励起色素レーザーを励起する。パルス幅の狭い励起光を用いることにより、フーリエ変換限界の色素レーザーパルスを得ることができる。色素レーザーの出力パルスはそのままでは時間分解ラマン分光の用途にはエネルギーが小さすぎるので、約104倍に増幅する。この増幅をkHz以上の高繰り返しで行うことが、時間分解ラマン分光の光源として極めて重要なポイントである。図2のシステムでは、モード同期Nd:YAGレーザーの出力の一部をシードしたNd:YAG再生増幅器を用い、その出力の倍波で色素増幅器を励起している。色素増幅器から得られる出力パルスは、波長588nm、繰り返し率2kHz、平均出力10〜30mWであり、上述のようにパルス幅3.2ps、スペクトル幅3.5cm-1の特性を持つ。この出力の一部を非線形光学結晶(BBO)により紫外光に変換する。得られた波長294nmの紫外パルス光を励起用(ポンプ)パルスとして用いる。紫外光に変換されなかった残りの可視パルス光をラマン検索用(プローブ)パルスとして用いる。ポンプパルスとプローブパルスの相互相関時間は4.1psで、Tailor基準に基づく時間分解能は2.2psである。ポンプパルスとプローブパルスの間の遅延時間は、プローブ光の光路に挿入した光学遅延回路によって調節される。適当な遅延時間が設定されたポンプ光とプローブ光はダイクロイックミラーで同軸状に重ね合わされ、レンズで集光された後、試料に照射される。ラマン散乱光は、レイリー散乱除去用の超狭帯域除去フィルターとシングル分光器により分光され、CCD検出器によって検出される。分光器のスループットとCCD検出器の量子効率から考えて、この検出系の感度はほぼ極限に近いレベルにあり、現在の感度をさらに数倍増大させることは原理的に不可能である。その意味で、図2のピコ秒時間分解ラマン分光装置は、時間分解能、波数分解能、検出感度の点でほぼ究極の性能を有していると言える。
現在、ピコ秒時間分解ラマン分光技術の発展は、より安定なTi:サファイアレーザー/再生増幅器の導入と、種々の非線形光学効果による波長領域の拡張の方向に向かっている8) 9)。FT型の分光計を用いたピコ秒ラマン測定の試みも報告されているが10) 11)、その実用性に関しては、今後の検討が必要である。また、Coherent Anti-Stokes Raman Scattering(CARS)を用いたピコ時間分解ラマン分光も盛んに行われている12) 13)。Taharaらが開発したピコ秒2次元マルチプレクスCARS法は、蛍光性の励起分子の時間分解ラマン測定に極めて有効であることが示された14) 15)。
3.3 応用
ピコ秒時間分解ラマン分光が実用化されてからまだ日が浅く、これまでのところまとまった成果と言えるのものはそう多くはない。既存の総説16)〜19)との重複を避けるために、ここでは溶液中のピコ秒振動緩和の研究に限ってやや詳しく解説する。
光励起分子の振動冷却過程の解析から、溶質/溶媒相互作用の詳細が明らかになりつつある。S1trans-スチルベンのC=Cニ重結合バンドが、光励起後数10psの時間領域で顕著な波数シフトを示すことが以前より知られており、余剰振動エネルギーを持った"ホット"なS1trans-スチルベンの冷却過程に対応するものと考えられていた16)。最近Iwataらは、ストークス/アンチストークス強度比とラマンスペクトルの温度変化から、このスペクトル変化が確かに振動冷却過程に対応するものであることを証明した。つぎに、さまざまな溶媒中における振動冷却過程を熱拡散方程式に基づき定量的に解析した20) 21)。その結果、光励起によってS1trans-スチルベン分子に与えられた余剰振動エネルギーは、励起後3ps以内に第一配位圏の溶媒分子によってまず共有され、次に溶媒の熱拡散定数で決まる時定数(10から15ps)でバルクの溶媒に散逸して行くという描像が得られた(図3)。
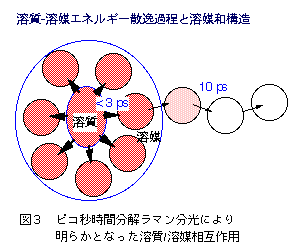
図3
さらに、励起直後における温度上昇の大きさから、溶媒がクロロホルムの場合、第一配位圏にある溶媒分子の数は5(振動-振動、振動-回転、振動-並進のエネルギー移動を考慮したとき)ないし12(振動-回転、振動-並進のエネルギー移動のみを考慮したとき)であることが示された。溶質/溶媒相互作用と、それに基づくエネルギーの流れの詳細が、このような形で明らかとなったのは、筆者の知る限り初めてのことである。S1trans-スチルベンの振動ダイナミクスに関しては上記以外にも様々な観点からの研究が報告されている22)〜29)。またS1trans-スチルベン以外の分子についても溶液中の振動緩和過程が調べられている30)〜32)。
4 ナノ・マイクロ秒時間分解赤外分光とその応用
4.1 装置および実験法
図4は分散型ナノ秒時間分解赤外分光計のブロック図である5)。高輝度MoSi2光源から射出される白色赤外光を、一対の楕円面鏡からなるビームコンデンサーにより約2mmに集光し、試料に入射させる。同時に、赤外光のスポットと重なるようにナノ秒紫外パルスレーザー光を照射し、試料を光励起する。試料を透過した赤外光は分散型赤外分光器によって単色化され、MCT(水銀-カドミウム-テルル)半導体検出器によって検出される。MCT検出器の出力は、ac結合された超低雑音前置増幅器により増幅された後、デジタルオシロスコープに記録される。分光器の掃引の各ステップごとにデジタルオシロスコープの波形を記録すると、掃引終了時には赤外透過強度変化の波数・時間の2次元データが得られる。この装置の特徴は、前置増幅器をac結合させることによって、透過赤外光強度の変化分のみを検出していることである。この方式によって、プリアンプのダイナミックレンジをフルに活用することができて、結果として吸光度変化(dA/A)にして10-6と、通常のFT-IRに比べて2桁高い検出感度が実現されている。時間分解能は、用いる検出器に依存するが、光起電力型のMCT検出器の場合、約50nsである。
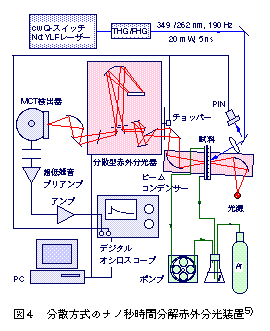
図4
同様のac結合方式は、ステップ掃引型FT-IR分光計にも適用できる。したがって原理的には、ステップ掃引FT方式は分散方式と同等の時間分解能と感度を持つことができるはずである。しかし、これまで得られたデータに関しては質、量とも分散方式がステップ掃引FT方式を凌駕している。最近、ステップ掃引FT方式の報告が増加しているので、今後の展開が注目されるところである。いずれにせよ、目的に応じて分散方式とステップ掃引FT方式を使い分けることが必要であろう。また赤外ダイオードレーザーを用いた気相の短寿命種の高分解測定33)や表面吸着種の時間分解赤外測定34)なども報告され始めている。
4.2 応用
4.2.1 化学的応用
すでに数多くの短寿命反応中間体が時間分解赤外分光法により検出・同定され、化学反応機構の議論をより実体を伴ったものにしている。伝統的に、金属カルボニル化合物を始めとする有機金属錯体の励起状態やラジカルの研究が最も盛んである。これはカルボニル基の赤外吸収の強度が極めて大きく、結果として実験が容易であることによる。この分野の文献の数は膨大であるので、総説をいくつかあげておくに止める35)〜37)。最近になって、溶液中の有機分子の励起状態38)〜40)、ラジカル41)〜44)互変異性体45)〜47)などの赤外スペクトルが続々と報告されるようになった。物理化学的なモデル系ではなく、フラスコの中でおきる実際の化学反応のプロセスを時間分解赤外分光によって直接に見ようとする試みが、今後一層増加して行くものと思われる。また表面吸着種への応用48)49)も、表面反応機構や触媒の研究との関連で、今後大きな展開を見せると期待される化学的応用の分野である。
4.2.2 生物学的応用
時間分解赤外分光は、時間分解ラマン分光と異なり、検索光として強いレーザー光を用いる必要がないので、光にたいして敏感な生体試料の測定に適している。バクテリオロドプシンの光化学反応サイクルは、さまざまな時間分解分光手法による広範な研究の対象となって来た。現在では、クロモフォアのシス-トランス異性化を含むフェムト/ピコ秒の初期過程がほぼ解明され、焦点は蛋白部分の構造変化をともなうナノ/マイクロ/ミリ秒の時間領域に移って来ている50)〜52)。バクテリオロドプシンの機能である光プロトンポンプを支配する蛋白質の構造変化を直接に捉える手法として、時間分解赤外分光が最も強力であると考えられている。光生物学的応用として他に、光合成反応中心53)54)、ATPの光放出55)、カルボキシヘモグロビンの光解離・再結合56)などが研究されている。光励起ではなく、レーザー照射による温度ジャンプにより、蛋白質の巻戻し過程を時間分解赤外分光で追跡する実験も報告されている57)。温度ジャンプだけでなく、pHジャンプなどの手法と組み合わせれば、光の関与しない生物現象の動的過程を、時間分解分赤外光によって調べる道が開ける。今後の発展が待たれる。
4.2.3 液晶の電場応答
外部電場に対する液晶の応答ダイナミクスの解明は、機能素子としての液晶セルの設計、製作、評価に関する極めて重要な情報を与える。従来、液晶の電場応答の研究では、液晶分子は剛体としてモデル化され、分子全体としての配向運動のみが考慮されていた。しかし、実際の液晶分子は必ず剛直な部位と柔軟な部位から構成されているので、それらが常に同一の動きをするという保証はどこにもない。時間分解赤外分光を用いれば、剛直部と柔軟部の電場応答ダイナミクスを、それぞれに対応する赤外吸収バンドの時間分解測定により、個別に観測することが可能となる。このような観点から、ネマチック液晶や強誘電性液晶について、時間分解赤外分光による研究が盛んに行われている。系の複雑さもあり、どの研究もまだ明確な結論を得るには至っていないが、今後大きく発展をする可能性が高い興味ある分野であるので、総説と原著論文をいくつか挙げておく58)〜66)。
5. ピコ秒時間分解赤外分光の現状
ピコ秒時間分解赤外分光は未だ揺藍期にあり、測定技術が確立されていない状況にある。多くの研究者がそれぞれ異なる手法に挑戦し、少数の測定例をデモンストレーションしているのが現状である。最近、Ti:サファイアレーザー/パラメトリック発振・増幅器と差周波発生法により比較的容易に赤外領域の超短パルスが得られるようになって来たので、今後研究例が急増するものと予測される。ここでは、総説67)と最近の注目される研究成果68)〜70)を幾つか挙げておく。
6. おわりに
多くの新しい分光手法がそうであったように、時間分解振動分光もまず物理化学の研究対象として登場し、10数年の発展の過程を経て、今ようやく活躍の舞台を分析・無機・有機化学へ移そうとしている。とくに、4.1で紹介した分散型ナノ秒時間分解赤外分光装置は、市販品が米国および日本の有機化学の研究室に納入され、有機化学者たちによって日常的に使われている。時間分解赤外分光が有機化学の標準的研究手段として認知される日もそう遠くはないものと思われる。一方、ピコ時間分解振動分光は、しばらくは物理化学の研究分野にとどまる、というのが筆者の予測である。しかし、未来が我々に何を用意してくれているのかを見通すことはもちろん不可能であり、ある日思いがけない応用の道が開け、ピコ秒の領域の振動分光が急速な展開を見る可能性を否定することはできない。もしそのような予期しない展開が、本稿の読者によってもたらされることになれば、筆者にとっては無上の喜びである。
文献
- E. B. Baker, C. D. Robb: Rev.Sci.Instrum.,14,362 (1943).
- P. Pagsberg, R. Wilbrandt, K. B. Hansen, K. V. Weisberg: Chem.Phys.Lett., 39, 538 (1976).
- H. Hamaguchi: in "Vibrational Spectra andStructure", Edited by J.R.Durig, Vol.16,p.227(1987), (Elsevier,Amsterdam).
- 加藤千尋、岩田耕一、濵口宏夫: 分光研究、40,255(1991).
- T. Yuzawa, C. Kato, M. W. George, H. Hamaguchi: Appl. Spectrosc. 48, 684 (1994).
- O. Weidlich, F. Siebert: Appl. Spectrosc.,47, 1394 (1993).
- K. Iwata, S. Yamaguchi, H. Hamaguchi: Rev.Sci.Instrum., 64, 2140 (1993).
- Y. Uesugi, Y. Mizutani, T. Kitagawa: Rev.Sci.Instrum., 68, 4001 (1997).
- M. Towire, A. W. Parker, W. Shaikh, P. Matousek: Meas.Sci.Technol., 9, 816 (1998).
- G. Jas, C. Wan, C. K Johnson: Appl.Spectrosc., 49,645 (1995).
- A. Sakamoto, H. Okamoto, M. Tasumi: Appl.Spectrosc., 52, 76 (1998).
- O. Weidlich, L. Uji, F. Jaeger, G. H. Atkinson: Biophys. J., 72, 2329 (1997).
- M. Schmitt, G. Knopp, A. Materny, W. Kiefer: J.Phys.Chem. A, 102, 4059 (1998).
- T. Tahara, H. Hamaguchi: Rev.Sci.Instrum.65, 3332 (1994).
- T. Ishibashi, H. Hamaguchi: J.Chem.Phys.,102, 2263 (1998).
- H. Hamaguchi, T. L. Gustafson: Annu.Rev.Phys.Chem. 45, 593 (1994).
- 岩田耕一、濵口宏夫:分光研究、44, 61 (1995).
- P. A. Thompson, R. A. Mathies: Tech.Chem.(N. Y.), 23, 185 (1995).
- 北川禎三:季刊化学総説、24, 2 (1995).
- K. Iwata, H. Hamaguchi: J.Mol.Liq.,65/66, 417 (1995).
- K. Iwata, H. Hamaguchi: J.Phys.Chem.,101,632 (1997).
- H. Hamaguchi, K. Iwata: Chem.Phys.Lett.,208, 465 (1993).
- A. P. Shkurinov, N. I. Koroteev, G. Jonusauskas, C. Rulliere: Chem.Phys.Lett.223, 573 (1994).
- J. Qian, S. L. Schultz, G. R. Bradburn,J. M. Jean: J. Lumin. 60/61, 727(1994).
- J. Qian, S. Schultz, J. M. Jean: Chem.Phys.Lett. 233, 9(1995).
- P. Matousek, A. W. Parker, W. T. Toner, M. Towire, D. L. A. deFaria, R. E. Hester, J. N. Moore: Chem.Phys.Lett. 237, 373 (1995).
- V. Deckert, K. Iwata, H. Hamaguchi: J.Photochem. Photobiol., A 102, 35 (1996).
- S. L. Shultz, J. Qian, J. M. Jean: J.Phys.Chem. A, 101, 1000 (1997).
- T. Nakabayashi, H. Okamoto, M. Tasumi: J.Phys.Chem. A, 101, 7189 (1997).
- S. Sato, T. Kitagawa: Springer Proc.Phys.,74, 104 (1994).
- S.-B. Ko, S.-C. Yu, J.B. Hopkins: Bull.KoreanChem.Soc., 15, 762 (1994).
- T. Nakabayashi, H. Okamoto, M. Tasumi: J.Phys.Chem.A, 101, 3494 (1997).
- K. Tanaka, T. Harada, K. Sakaguchi, K. Harada,T. Tanaka: J. Chem.Phys., 103, 6450 (1995).
- E. Borguet, H.-L. Dai: Adv.Ser.Phys.Chem.,5, Pt. 1, 243 (1995).
- J. J. Turner, M. W. George, M. Poliakoff: Spec.Publ.-R.Soc. Chem., 163, 13 (1995).
- J. R. Schoonover, G. F. Strause, K. M. Omberg,R. B. Dyer: Comments Inorg. Chem., 18, 165(1996).
- P. C. Ford, J. S. Bridgewater, B. Lee: Photochem. Photobiol. 65, 57 (1997).
- M. Hashimoto, H. Hamaguchi: J.Phys.Chem:99, 7875 (1995).
- T. Yuzawa, H. Hamaguchi: J.Mol.Struct.,352, 489 (1995).
- H. Sun, H. Frei: J.Phys.Chem.B, 101,205 (1997).
- C. E. Brown, A. G. Neville, D. M.Rayner,K. U. Ingold: Aust. J. Chem., 48, 363 (1995).
- K. Iwata, H. Hamaguchi: J.Mol.Struct.,413-4, 101 (1997).
- A. Barth, J. E. T. Corrie, M. J. Gradwell,Y. Maeda, W. Maentele, T. Meier, D. R. Trentham: J.Am.Chem.Soc., 119, 4149 (1997).
- S. Srivastava, J. P. Toscano, R. J. Moran,D. E. Falvey: J. Am. Chem. Soc., 119, 11552(1997).
- M. Itoh, T. Yuzawa, H. Mukaihata, H. Hamaguchi:Chem.Phys.Lett., 233, 550 (1995).
- K. Tokumura, M. Natsume, T. Nakagawa, M. Hashimoto, T. Yuzawa, H. Hamaguchi, M. Itoh: Chem.Phys.Lett., 271, 320 (1997).
- K. Kobayashi, M. Iguchi, T. Imakubo, K. Iwata,H. Hamaguchi: J.Chem.Soc.,Chem.Commun.,763 (1998).
- J. Huberty, R. J. Madix: Surf.Sci., 334,77 (1995).
- A. Yamakata, J. Kubota, J. N. Kondo, C. Hirose, K. Domen, F. Wakabayashi, K. Tamaru: J.Phys.Chem.B, 102, 4401 (1998).
- J. Sakai, T. Yuzawa, H. Kandori, A. Maeda,H. Hamaguchi: Biophys. J., 68, 2073 (1995).
- O. Weidlich, N. Friedman, M. Sheves, F. Siebert: Biochemistry, 34, 13502 (1995).
- C. Zscherp, J. Heberle: J.Phys.Chem B,101, 10542 (1997).
- R. Hienerwadel, S. Grzybek, C. Fogel, W. Kreutz, M. Y. Okamura, M. L. Paddock, J. Breton, E. Nabedryk, W. Maentele: Biochemistry, 34, 2832 (1995).
- H. Zhang, G. Fischer, T. Wydrzynski: Biochemistry, 37, 5511 (1998).
- J. Backmann, H. Fabian, D. Naumann: FEBS Lett., 364, 175 (1995).
- A. Barth, K. Hauser, W. Maentele, J. E. T. Corrie, D. Tretham: J. Am. Chem. Soc.,117, 10311 (1995).
- X. Hu, H. Frei, T. G. Spiro: Biochemistry,35, 13001 (1996).
- 浦野妙子、濵口宏夫:化学、49、70(1994).
- T. I. Urano, H. Hamaguchi: Appl.Spectrosc.47, 2108 (1993).
- S. V. Shilov, S. Okretic, H. W. Siesler: Vib.Spectrosc. 9, 57 (1995).
- N. Katayama, T. Sato, Y. Ozaki, K. Murashiro,M. Kikuchi, S. Saito, D. Demus, T. Yuzawa,H. Hamaguchi: Appl.Spectrosc.,49, 977 (1995).
- H. Toriumi: Mol.Cryst.Liq.Cryst.Sci.Technol., Sect.A, 262, 1659 (1995).
- M. Czarnecki, N. Katayama, M. Satoh, T. Watanabe, Y. Ozaki: J.Phys.Chem., 99, 14101 (1995).
- S. V. Shilov, H. Skupin, F. Kremer, T. Wittig,R. Zentel: Phys.Rev.Lett., 79, 1686 (1997).
- A. L. Verma, B. Zhao, S. M. Jiang, J. C. Sheng, Y. Ozaki: Phys. Rev.E,56, 3053 (1997).
- H. Toriumi, T. Akahane: Jpn.J.Appl.Phys.,Part1, 37, 608 (1998).
- B. D. Locke, R. M. Hochstrasser : Adv. Spectrosc., 21, 1 (1993).
- M. W. George, T. P. Dougherty, E. J. Heilweil : J. Phys. Chem., 100, 201 (1996).
- B. D. Locke, R. M. Hochstrasser: Advances in Spectroscopy, 21, 1 (1993).
- M. W. George, T. P. Dougherty, E. J. Heilweil: J.Phys.Chem., 100, 201 (1996).
- R. Laenen, C. Rauscher: Chem.Phys Lett.,274, 63 (1997).
- H. Okamoto: Chem.Phys.Lett., 283, 33 (1998).